biomod2パッケージを使うときのメモ
Rの種分布モデリングのためのbiomod2パッケージを使うときのメモです。 新しめのパッケージということもあり、ドキュメントが少なく、使い方がわかりにくい部分がありました。 また、バグと思われる部分もありました。
あと、モデリングの際に警告(warnings)が表示されることが多いです。 それらについて、メモを残しておきます。
種名について
BIOMOD_FormatingData() 関数では、resp.name 引数で種名を指定します。 この時、種名がアンダースコア _ を含むと、勝手にピリオド . に変換されます。
例えば、"My_Species" と指定すると、"My.Species" に変換されます。
この時、以下のような出力がされます。
> myBiomodData <- BIOMOD_FormatingData(
+ resp.var = myResp,
+ expl.var = myExpl,
+ data.type = "binary",
+ dir.name = dir_models,
+ resp.xy = myRespXY,
+ resp.name = myRespName
+ )
-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-= Aesculus_turbinata Data Formating -=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=
! Response variable name was converted into Aesculus.turbinata
! Some NAs have been automatically removed from your data
-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-= Done -=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=-=理由はわかりません。 個人的には ピリオド . 区切りよりも アンダースコア _ 区切りの方が好きなので、これは不便でした。 実害は特にないです。
モデリングオプション
モデリングオプションの設定が厄介でした。
GAM
GAMを使う場合、models にそのまま “GAM” と指定すると、以下のような警告が出ました。
GAMを使う場合、どのパッケージのGAMを使用するかを明示的に指定することで、警告が出なくなりました。 例えば、在不在データですべてのモデルを使用する際は、以下のようになります。 bm_ModelingOptions() の引数は最小限だけ記載しています。
models <- c(
"ANN",
"CTA",
"DNN",
"FDA",
"GAM.mgcv.gam",
"GBM",
"GLM",
"MARS",
"MAXENT",
"MAXNET",
"RF",
"RFd",
"SRE",
"XGBOOST"
)
options <- bm_ModelingOptions(
data.type = "binary",
models = models,
strategy = "user.defined"
)"GAM.mgcv.gam" とすることで、mgcv パッケージの gam() 関数を明示的にしていし、警告が出ないようにします。
しかし、そのあとの bm_Modeling() の models 引数にそのまま先ほど指定した models を指定して実行すると、以下のようなエラーが出ました。
Error in `.fun_testIfIn()`:
!
models with binary data type must be 'ANN', 'CTA', 'DNN', 'FDA', 'GAM', 'GBM', 'GLM', 'MARS', 'MAXENT', 'MAXNET', 'RF', 'RFd', 'SRE' or 'XGBOOST
Hide Traceback
▆
1. └─biomod2::BIOMOD_Modeling(...)
2. └─biomod2:::.BIOMOD_Modeling.check.args(...) at biomod2/R/BIOMOD_Modeling.R:408:3
3. └─biomod2:::.fun_testIfIn(...) at biomod2/R/BIOMOD_Modeling.R:620:3つまり、bm_Modeling() の models 引数には、"GAM" と指定する必要があるようです。 bm_ModelingOptions() の後に、以下のように models を書き換えます。
models[models == "GAM.mgcv.gam"] <- "GAM"その後、bm_Modeling() の models 引数に models を指定して実行すると、エラーが出ずに実行できました。 二度手間のような気もしますが、仕方ないようです。
警告が気にならない方は、bm_ModelingOptions() の models 引数に "GAM" と指定しても動作します。 この場合、mgcv パッケージの gam() 関数が使用されます。
MAXENT を使うときの注意点
MAXENT だけはRパッケージではなく、ソフトウェア経由から利用しています。 すこし設定が必要です。 そのため、Javaが必要です。
Java のインストール
コマンドプロンプトやPowerShellで java -version を実行して、Javaがインストールされていることを確認してください。 インストールがされていない場合は、以下のように表示されると思います。
'java' は、内部コマンドまたは外部コマンド、
操作可能なプログラムまたはバッチ ファイルとして認識されていません。Java のインストールはいくつか方法があります。 よく目にするのは公式の Oracle のものですが、個人利用であれば Adoptium のものを使うのが簡単で便利だと思います。
JDK か JRE のどちらでも問題はないですが、JRE の方が軽量です。 JDK は開発用のツールも含まれているため、サイズが大きいです。 私は JDK をインストールしました。
バージョンは 8 以降であれば問題ないと思います。 私は 8 にしました。 ちなみに、Maxent の最新版は v3.4.3 で、こちらは2020年リリースです。 新しすぎる JAVA だと不具合が生じる可能性があるので、あまり新しすぎないバージョンを選ぶのが無難かもしれません。
Adoptium の Temurin® JDK をインストールするには、CLI(コマンドラインインターフェース) がもっとも簡単です。
インストールガイドページ に OS ごとのインストール方法が書かれています。
Windows であれば、パッケージマネージャーの WinGet を使うのが簡単です。
下記のコマンドでインストールしました。
winget install EclipseAdoptium.Temurin.8.JDK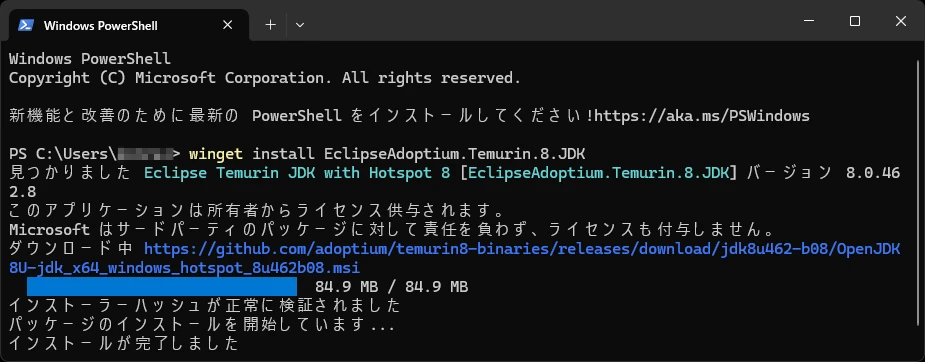
インストールが完了したら、コマンドプロンプトやPowerShellを再起動し、java -version を実行して、インストールが成功したことを確認します。
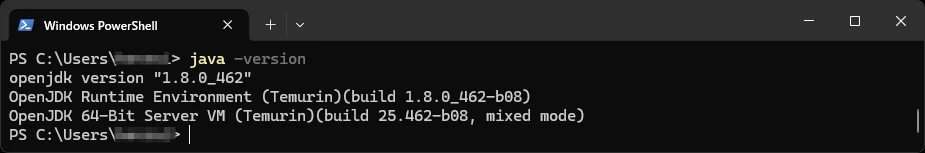
これで PC に Java がインストールされました。 次に、R から Java を使えるように設定します。
設定アプリを開き、検索で 「環境変数」と入力し、「システム環境変数の編集」を選択します。
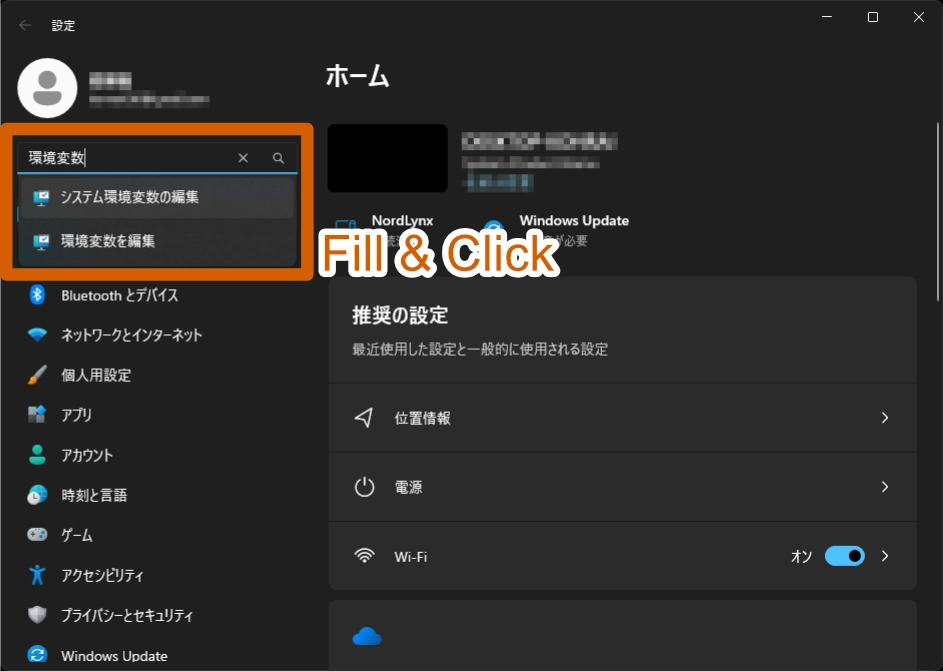
[システムのプロパティ] ウインドウが開くので、「環境変数(N)…」ボタンをクリックします。
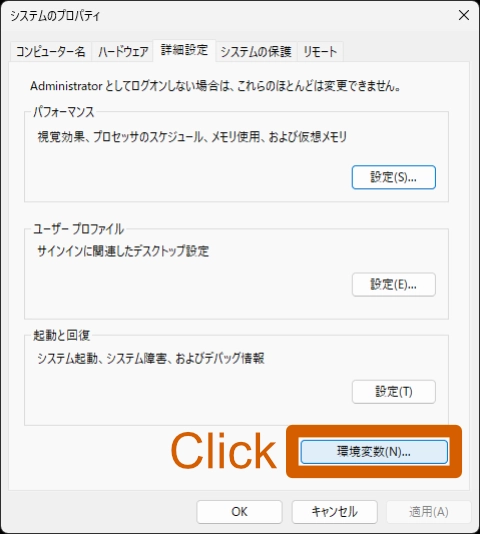
[環境変数] ウインドウが開きます、 [システム環境変数] に JAVA_HOME という名前の変数があるかを確認します。 次に、変数から [Path] を選択し、「編集(I)…」ボタンをクリックします。
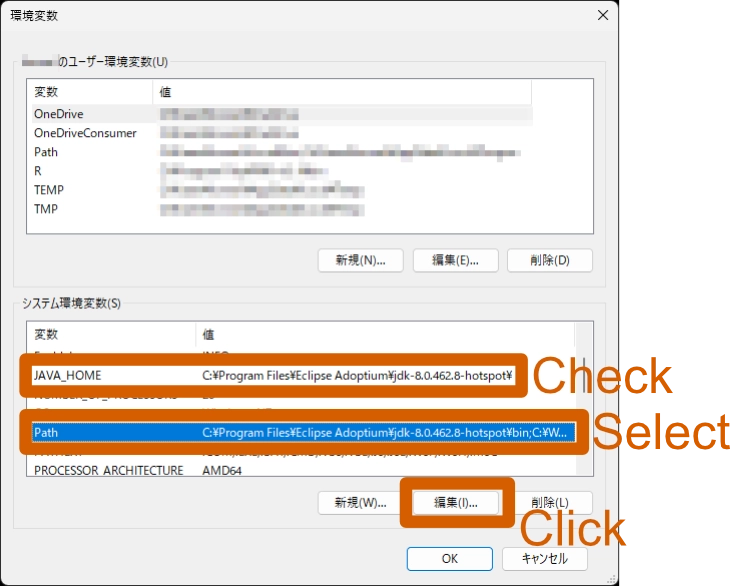
[環境変数名の編集] ウインドウが開きます。 「新規(N)」 ボタンをクリックし、新しい行に %JAVA_HOME%\bin と入力します。 [OK] をクリックし、すべてのウインドウを閉じます。
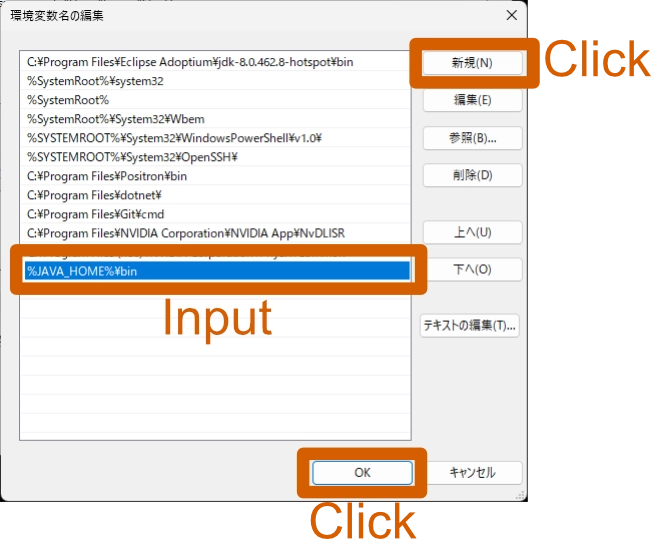
PC を再起動し、Sys.which("java") と入力します。 R のセッションの再起動だけではダメでした。
Path の設定が正しくできている場合は、Java のパスが表示されます。 私の環境では以下のように表示されました。
> Sys.which("java")
java
"C:\\PROGRA~1\\ECLIPS~1\\JDK-80~1.8-H\\bin\\java.exe" ちなみに、Java がインストールされいなかったり、Path が通っていない PC で R から Maxent を実行しようとすると、以下のようなエラーが出ます。
Error in system(command = maxent.command, wait = TRUE, intern = TRUE) :
'java' not found
In addition: There were 16 warnings (use warnings() to see them)Maxent のダウンロード
Maxent のソフトウェアは、以下からダウンロードできます。
maxent.jar ファイルの配置
また、MAXENTのソフトウェアをダウンロードして、Rから参照できるようにする必要があります。
Warning message:
In .BIOMOD.options.default.check.args(mod, typ, pkg, fun) :
'maxent.jar' file is missing in current working directory (C:/Users/konrai/Dropbox/doctor-research).
It must be downloaded (https://biodiversityinformatics.amnh.org/open_source/maxent/) and put in the working directory.それでも、bm_ModelingOptions() 関数での単一モデリングの間に、以下のように出力されてしまいました。
-=-=-=--=-=-=- <myRespName>_allData_RUN1_MAXENT
Creating Maxent Temp Proj Data...
Getting predictions...
Getting predictor contributions...
Error in system(command = maxent.command, wait = TRUE, intern = TRUE) :
'java' not found
In addition: There were 17 warnings (use warnings() to see them)どうやら、RからJavaが見つけられないようです。
モデリングオプションを反映させる
bm_ModelingOptions() 関数で設定したモデリングオプションは、bm_Modeling() 関数に options 引数で渡す必要があります。 この際もやや引数の指定がややこしいです。
並列化について
単一モデリングの際の BIOMOD_Modeling() 関数では、nb.cpu 引数で並列化のためのCPUコア数を指定できます。 デフォルトでは 1 ですが、例えば 2 と指定すると、2コアを使用して並列化します。
しかし、ドキュメントには書いてありませんが、これはWindowsでは動作しません。 Windowsでは、nb.cpu 引数を指定しても無視され、常にシングルコアで実行されます。 LinuxやMacOSでは動作します。
ほかにモデリングを高速化する方法を考えています。
set_new_dirname() におけるバグ?
モデリングオブジェクトでのフォルダ名を変更する set_new_dirname() 関数にバグがあるようです。 しかも、biomod2 でのモデリングオブジェクトでは、パス名は絶対パスで保存されているため、ほかのPCではモデリングオブジェクトを読み込んでもおそらくそれ以降の解析ができないです。
例えば、bm_ModelingOptions() 関数で単一モデリングをしておいて、後で BIOMOD_EnsembleModeling() 関数で別の関数でアンサンブルモデルを構築したいとします。
そのような場合は、基準とするパスが異なってしまうため、途中でエラーが生じ、モデリングが止まります。
原因
関数のコードを見ていたら、以下の二つのことに気が付きます。 まだできていませんが、GitHubで既存の Issue があるか確認してみます。 なかったらIssue を立てるか、自分で修正して Pull Request を出すか検討したいと思います。
対処法(失敗)
対処法として、モデルを読み込んでから自分でオブジェクト内のパスを変更する方法を試しました。 しかし、結果はうまくいきませんでした。 関係するすべてのパスを変更してみたのですが、モデリング関数では、処理の途中でもう一回モデルをロードする処理が入っているようで、、その際に元のパスが使われてしまうようです。
それならばと、パスを変更した後、ローカルフォルダに上書き保存をしてみました。 しかし、これも失敗しました。 ファイル形式が特殊なようで、自分で保存しても、正しく保存されないようです。 もう一度読み込もうとしたらエラーが出ました。
BIOMOD_Modeling() における警告表示
ROC 作成の際の警告表示
BIOMOD_Modeling() 関数でモデリングを実行する際、metric.eval 引数に "AUCroc" を指定すると、以下のような警告が表示されました。
Warning messages:
1: In coords.roc(roc1, "best", ret = c("threshold", "sens", "spec"), :
'transpose=TRUE' is deprecated. Only 'transpose=FALSE' will be allowed in a future version.この関数の内部で、pROC::coords() 関数が使用されていて、transpose = TRUE と指定されているが、pROC パッケージの最新版では transpose = TRUE は非推奨になっているようです。
BIOMOD_Modeling() 関数からは制御できないため、警告を無視するしかないようです。 将来的にエラーになる可能性がありますが、現時点では問題ないようです。
macv::gam() に関する警告表示
BIOMOD_Modeling() 関数でモデリングを実行する際、mgcv::gam() でGAMを使用すると、以下のような警告が表示されました。
10: In newton(lsp = lsp, X = G$X, y = G$y, Eb = G$Eb, UrS = G$UrS, ... :
Iteration limit reached without full convergence - check carefullyこれはどうやら、mgcv::gam() 関数の内部で使用されている newton() 関数からの警告のようです。 mgcv::gam の内部最適化(ニュートン法)が最大反復回数に達して収束しなかった、という警告のらしいです。 データの当てはまりやスムーズの自由度設定が厳しすぎる場合に起きやすいとのことです。
対処法としては、平滑の基数 k を小さくする、select=TRUE、method="REML" などを検討する、などがあるようです。 しかし、BIOMOD_Modeling() 関数の引数では mgcv::gam() の引数を直接指定できないため、対処が難しいです。
そもそもの、説明変数の多重共線性や外れ値の問題があるかもしれません。
参考:
glm() に関する警告表示
BIOMOD_Modeling() 関数でモデリングを実行する際、glm() でGLMを使用すると、以下のような警告が表示されました。
46: glm.fit: algorithm did not converge
47: glm.fit: fitted probabilities numerically 0 or 1 occurred
48: the glm algorithm did not converge for response "種名"MAXENT に関する警告表示
21: In file.remove(m_predictFile) :
cannot remove file 'c:/Users/<アウトプットパス>/Predictions/Pred_swdBis_23498.csv', reason 'Permission denied'R 側でファイルを削除しようとしたが、権限がないため削除できなかった、という警告のようです。 以前に同じファイルが開かれている場合などに発生することがあるようです。 私の場合は、同じ種で複数回モデリングを実行した際に発生しました。
パスをDropboxにしていることが原因かもしれません。 ローカルフォルダに変更したら警告が消えるかもしれません。
並列化について
1: executing %dopar% sequentially: no parallel backend registered並列化のためのバックエンドが登録されていないため、並列化が無効になり、シーケンシャルに実行される、という警告のようです。
私は Windows を使っているため、nb.cpu 引数を指定しても無視され、常にシングルコアで実行されます。
この警告に関しては、無視して問題ないと思います。
38: In newton(lsp = lsp, X = G$X, y = G$y, Eb = G$Eb, UrS = G$UrS, ... :
Iteration limit reached without full convergence - check carefully
46: glm.fit: algorithm did not converge
47: glm.fit: fitted probabilities numerically 0 or 1 occurred
48: the glm algorithm did not converge for response "Aesculus.turbinata"